
Reformulating Chemistry for More Efficient Quantum Computation
March 22, 2018
Posted by Ryan Babbush, Senior Research Scientist, Quantum AI Team
The first known classical “computer” was the Antikythera mechanism, an analog machine used to simulate the classical mechanics governing dynamics of celestial bodies on an astronomical scale. Similarly, a major ambition of quantum computers is to simulate the quantum mechanics governing dynamics of particles on the atomic scale. These simulations are often classically intractable due to the complex quantum mechanics at play. Of particular interest is the simulation of electrons forming chemical bonds, which give rise to the properties of essentially all molecules, materials and chemical reactions.
 |
| Left: The first known computing device, the Antikythera mechanism: a classical machine used to simulate classical mechanics. Right: Google’s 22 Xmon qubit “foxtail” chip arranged in a bilinear array on a wafer, the predecessor to Google’s new Bristlecone quantum processor with 72 qubits, a quantum machine we intend to use to simulate quantum mechanics, among other applications. |
Today, we highlight two recent publications with theoretical advances that significantly reduce the cost of these quantum computations. Our results were presented at the Quantum Information Processing and IBM ThinkQ conferences.
The first of these works, “Low-Depth Quantum Simulation of Materials,” published this week in Physical Review X, was a collaboration between researchers at Google, the group of Professor Garnet Chan at Caltech and the QuArC group at Microsoft. Our fundamental advance was to realize that by changing how molecules are represented on quantum computers, we can greatly simplify the quantum circuits required to solve the problem. Specifically, we specially design basis sets so that the equations describing the system energies (i.e. the Hamiltonian) become more straightforward to express for quantum computation.
To do this, we focused on using basis sets related to functions (plane waves) used in classical electronic structure calculations to provide a periodic representation of the physical system. This enables one to go beyond the quantum simulation of single-molecules and instead use quantum computers to model realistic materials. For instance, instead of simulating a single lithium hydride molecule floating in free space, with our approach one can quantum simulate a crystal of lithium hydride, which is how the material appears in nature. With larger quantum computers one could study other important materials problems such as the degradation of battery cathodes, chemical reactions involving heterogeneous catalysts, or the unusual electrical properties of graphene and superconductors.
In “Quantum Simulation of Electronic Structure with Linear Depth and Connectivity,” published last week in Physical Review Letters with the same collaborators and a Google intern from the Aspuru-Guzik group at Harvard, we leverage the structure introduced in the work above to design algorithms for near-term quantum computers with qubits laid out in a linear array. Whereas past methods required such quantum computers to run for time scaling as the fifth power of the number of simulated electrons for each dynamic step, our improved algorithm runs for time scaling linearly with respect to the number of electrons. This reduction in computational cost makes it viable to perform quantum chemistry simulations on near-term devices with fewer gates in each quantum circuit, possibly avoiding the need for full error-correction.
Even with these improvements, it is no small task to deploy such new technology to outperform classical quantum chemistry algorithms and methods which have been refined in parallel with the development of classical computers for more than eighty years. However, at the current rate of advances in quantum algorithms and hardware, quantum technologies may provide chemists with an invaluable new tool. We look forward to sharing our research results as they develop.
Other posts of interest
-
April 19, 2024
Improving Gboard language models via private federated analytics- Algorithms & Theory ·
- Distributed Systems & Parallel Computing ·
- Product ·
- Security, Privacy and Abuse Prevention
-

April 16, 2024
Solving the minimum cut problem for undirected graphs- Algorithms & Theory
-
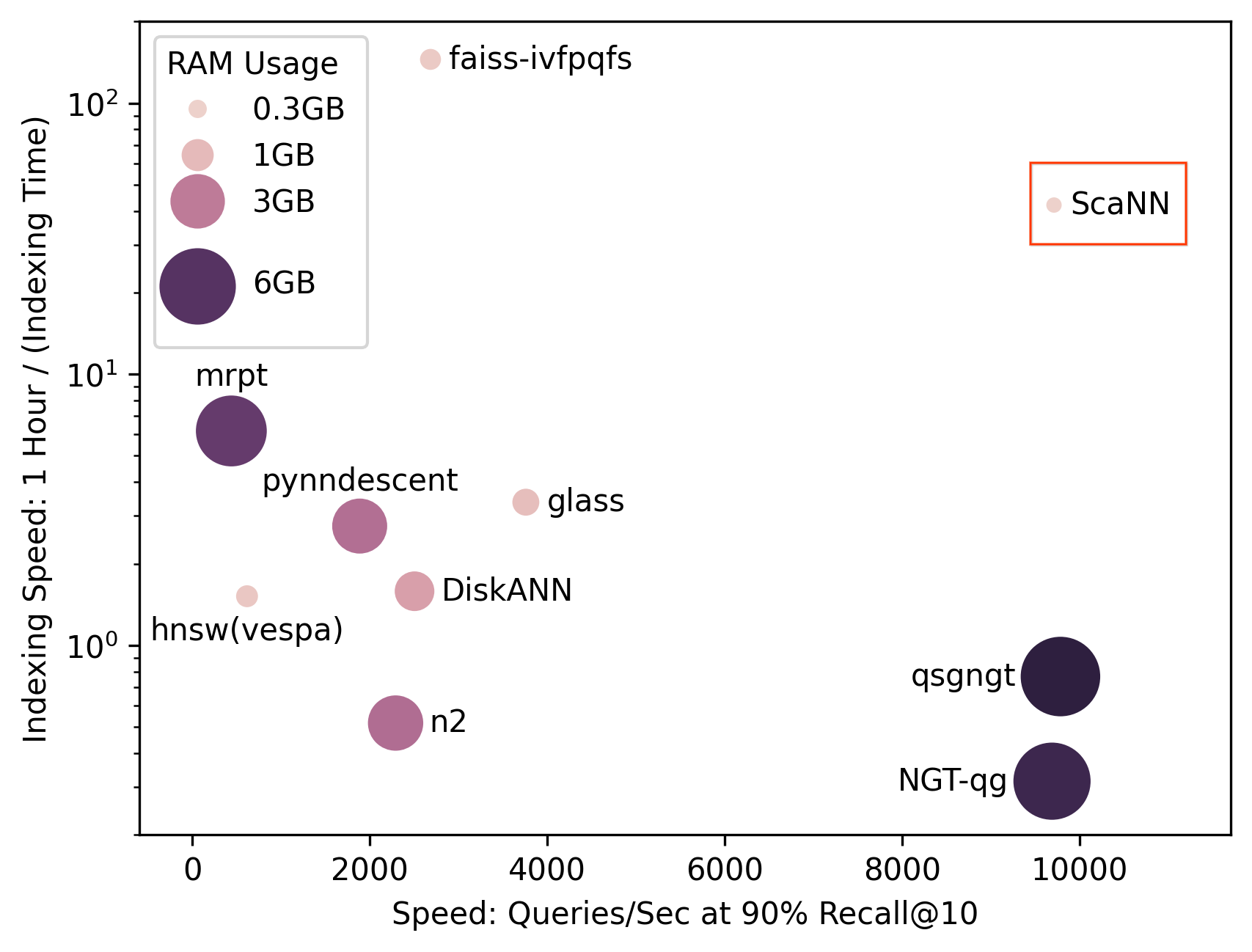
April 10, 2024
SOAR: New algorithms for even faster vector search with ScaNN- Algorithms & Theory ·
- Conferences & Events ·
- Data Mining & Modeling